Md Babul Hosen/iStock via Getty Images
Applied Therapeutics, Inc. (NASDAQ:APLT) is a promising clinical-stage biotech that focuses on treating symptoms of galactosemia in the central nervous system [CNS]. Galactosemia is rare when the body lacks a particular enzyme that metabolizes galactose. As a result, galactose and galacticol accumulate, leading to aberrant neuronal activity due to toxic levels of aldose reductase [AR] in cells. This can cascade into potentially serious adverse effects if left untreated, which is why APLT’s leading drug candidate, AT-007, is vital for this patient population. Moreover, I believe the company’s valuation multiple is reasonable in light of its ample resources and late-stage approval process, so I rate the stock a “strong buy” for investors aware of the inherent biotech risks.
Betting on AT-007: Business Overview
Applied Therapeutics is a clinical-stage biopharmaceutical company based in New York. Founded in 2016, APLT specializes in developing drug candidates against molecular targets and pathways that produce diseases with unmet medical needs. The company believes that applying its science-based development strategies can reduce the time from discovering medicines to their approval. Its leading drug candidate for treating galactosemia is rapidly nearing FDA approval in late 2024, which would unlock a significant TAM and transition the company toward commercialization efforts.
Source: APLT Corporate Overview – March 2024.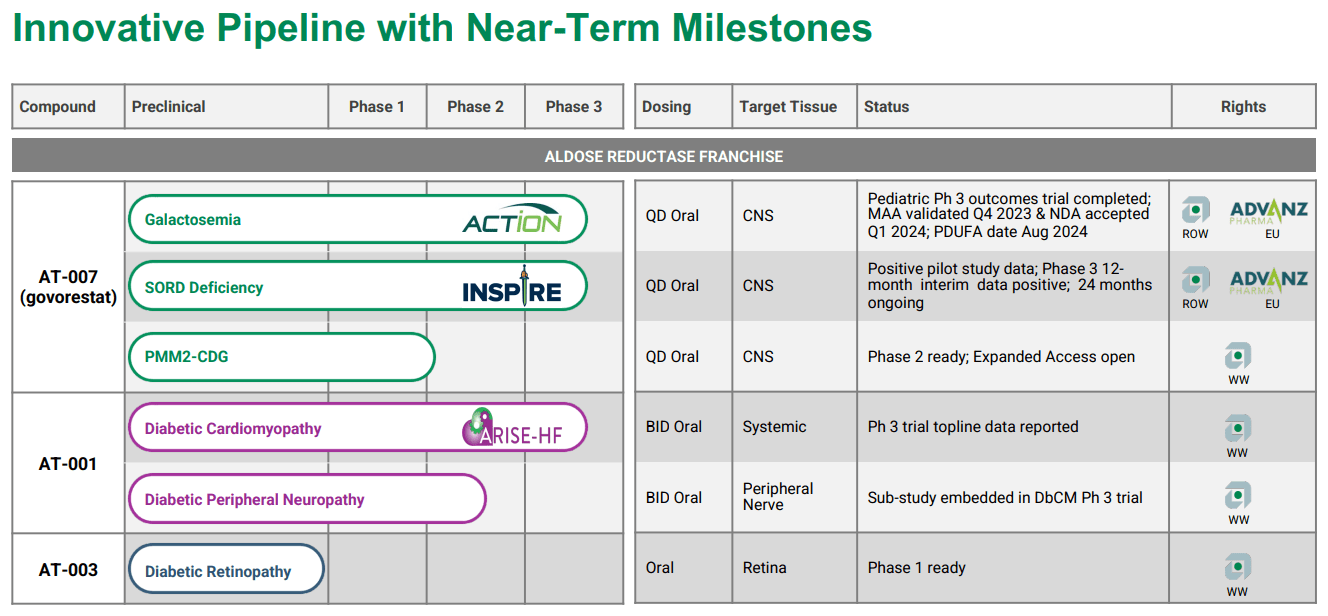
APLT’s underlying science starts with the Aldose Reductase Inhibitor [ARI] program. Here, the company designs and studies multiple compounds to fight against rare diseases such as Galactosemia, Sorbitol Dehydrogenase [SORD] Deficiency, and Phosphatomannomutase 2 Congenital Disorder of Glycosylation [PMM2-CDG]. Govorestat inhibits the enzyme aldose reductase, which allows the accumulation of the different toxic substances present in those conditions. The target tissue is the central nervous system [CNS], as these conditions can rapidly cascade into CNS problems if left untreated. For these illnesses, current treatment regimens only manage the symptoms and complications but do not address the root causes.
Concretely, galactosemia is a rare genetic disorder caused by a lack of the enzyme galactose-1-phosphate uridylyltransferase [GALT] for metabolizing galactose, a sugar component of lactose present in milk and dairy products. Galactose accumulates in the blood and can lead to serious complications like liver damage, cataracts, kidney failure, and even intellectual disability. Here’s where APLT’s Govorestat [AT-007] shines, as indicated for Galactosemia, progressing in phase 3 of its clinical trials. The pediatric phase 3 is completed, the Marketing Authorization Application [MAA] was validated in Q4 2023, the New Drug Application [NDA] was accepted in Q1 2024 under Priority Review, and the PDUFA date is November 28, 2024.
Source: APLT Corporate Overview – March 2024.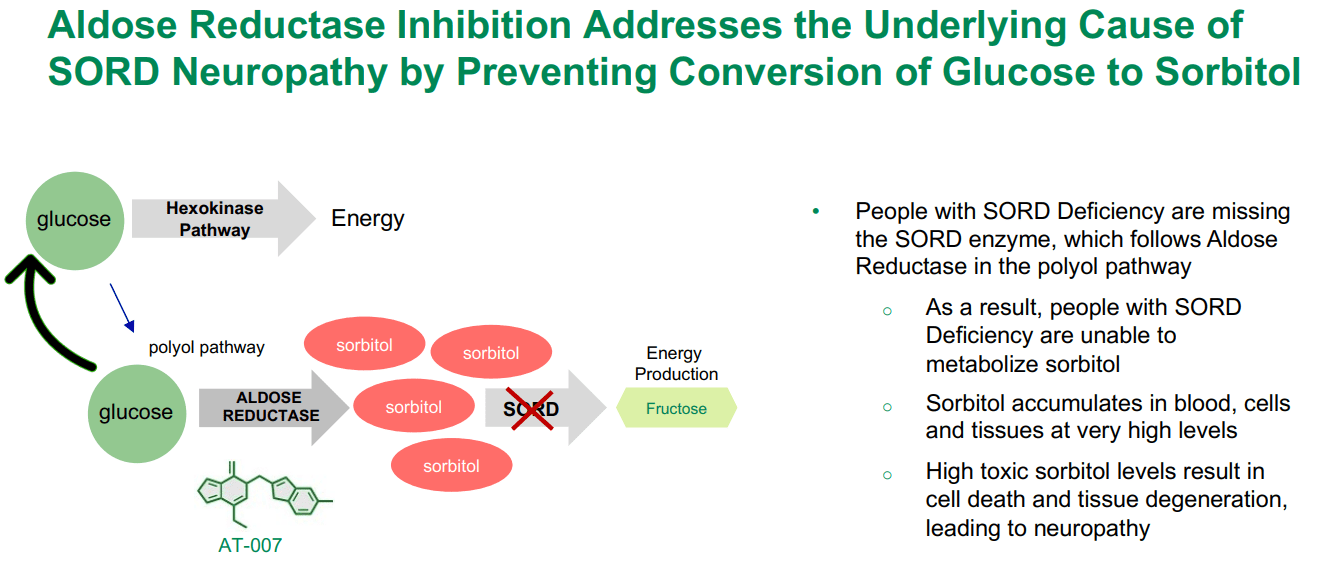
Interestingly, APLT’s AT-007 can also potentially address SORD Deficiency. This genetic illness presents with a deficiency of this enzyme, preventing the conversion of sorbitol into fructose. This causes an accumulation of sorbitol in nerve cells, which can cause peripheral neuropathy, muscle weakness, and pain. Govorestat, for this indication, is in phase 3. The action mechanism is similar, as it deals with the unwanted accumulation of sorbitol in neurons that lead to toxic patient outcomes. It’s worth mentioning that if AT-007 is approved for galactosemia, I believe it would corroborate its effectiveness, increasing the approval odds for related indications such as SORD, by extension.
Lastly, PMM2-CDG is also a rare genetic condition caused by a mutation in the PMM2 gene, which provokes a deficiency in the enzyme phosphomannomutase 2. The lack of this enzyme impacts the glycosylation process, which attaches sugar molecules to proteins and liquids for the body’s normal functioning. As a result, symptoms such as intellectual disability, liver dysfunction, and coagulation abnormalities, among others, appear. This is why AT-007 (Govorestat) can also have a potential indication for PMM2-DG, and APLT is initiating phase 2.
Govorestat has the European Medicines Agency’s [EMA] orphan medicinal product designation and the FDA’s Orphan Drug and Pediatric Rare Disease Designation for Galactosemia and PMM2-CDG.
Beyond AT-007: Company Updates
It’s also worth noting that APLT has another drug candidate in phase 3, Caficrestat [AT-001], for Diabetic Cardiomyopathy. For this indication, the target tissue is the peripheral nerve. Also, APLT has AT-003 in early phase 1 clinical studies for diabetic retinopathy. Its target tissue is the retina. However, I believe AT-007’s tangible near-term potential largely overshadows these potential drug candidates, so I focus on Govorestat as the company’s main value driver.
Furthermore, according to APLT’s Q1 2024 financial results, the NDA and MAA submissions delivered to the FDA and EMA for AT-007 include data from the phase 3 ACTION-Galactosemia Kids study in children aged 2-17 with Galactosemia, the phase 1/2 ACTION-Galactosemia study in adult patients with Galactosemia, and also preclinical data. The complete submissions highlight the efficacy and safety of the drug against Galactosemia for different age groups. Overall, it seems AT-007 is effective at reducing galacticol levels compared to placebo, with a reasonably safe tolerability profile.
Source: APLT Corporate Overview – March 2024.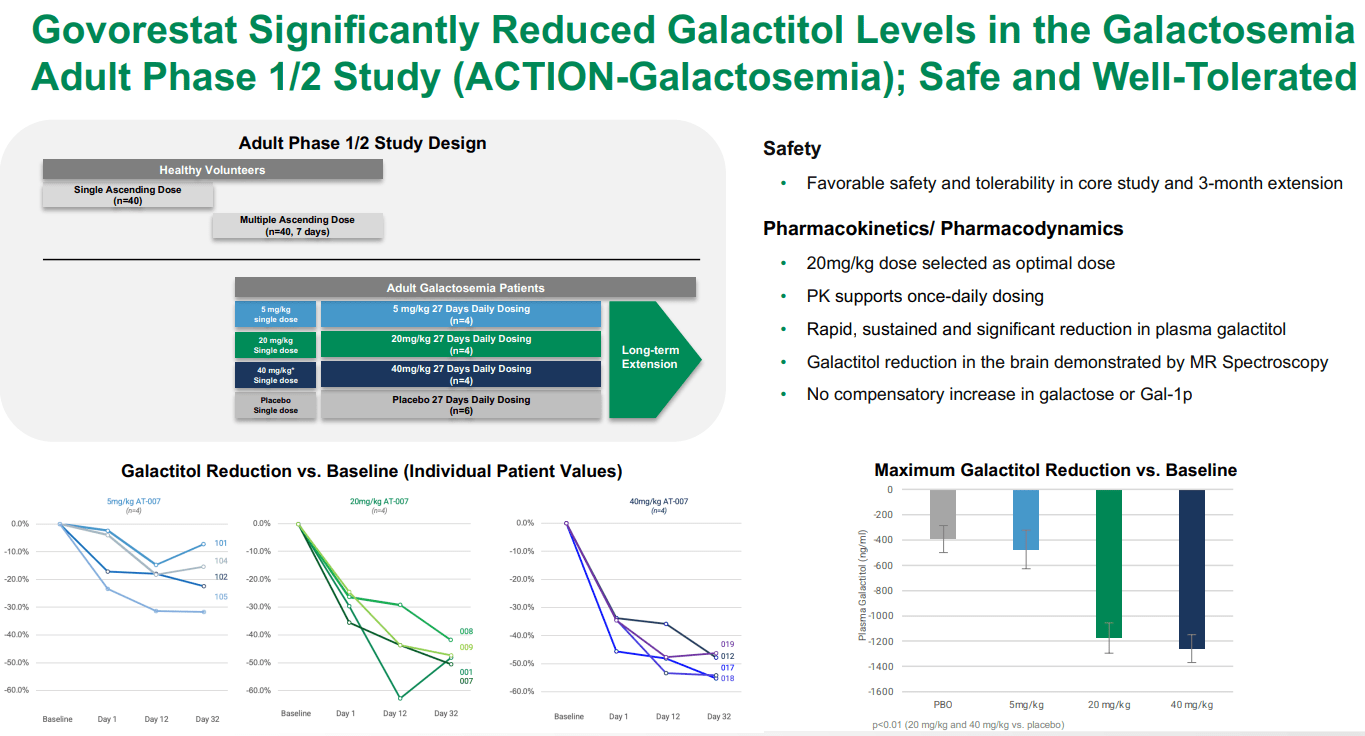
Additionally, the company presented results from its phase 3 ARISE-HF trial for AT-001 on diabetic cardiomyopathy patients. The data demonstrated stabilization of cardiac functional capacity compared to patients without treatment. The AT-001 cohort also showed no worsening symptoms or heart failure progression compared to the placebo group. These results suggest a promising potential as a viable possible cure for diabetic cardiomyopathy. So overall, I’d argue that APLT has made substantial progress across all of its IP portfolio and continues to advance its pipeline with positive clinical trial data.
Worth the Price: Valuation Analysis
From a valuation perspective, APLT trades at a modest market cap of $466.2 million. It currently holds $146.5 million in cash and equivalents against no debt (only operating lease obligations). This is mostly thanks to the recent equity offering that bolstered the company’s coffers by $100.0 million in February 2024. It’s also noteworthy that the company priced its shares at $7.00 for such an offering, and today, it trades at just $4.08 per share. So, while previous investors might have been affected by this, it implies a favorable price for new investors.
However, APLT hasn’t generated product revenues so far, but the company seems to be on track to generate its first product sales by early 2025. Assuming the FDA approves the PDUFA by November 2024, APLT could start generating revenues by early 2025, and if the EMA follows a similar timeline, then next year could be a pivotal year for the company. Seeking Alpha’s APLT dashboard forecasts the company will produce $45.7 million in sales by next year.
Source: Seeking Alpha.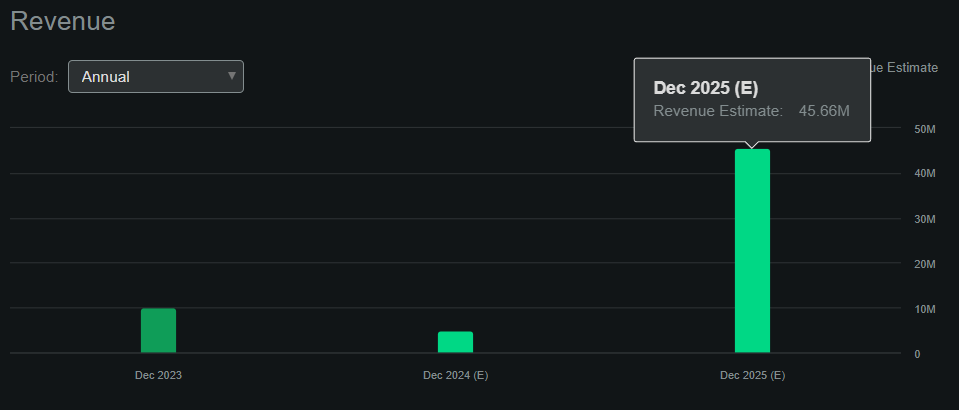
This means the stock trades at a forward P/S ratio of about 10.2, which is self-evidently high. In fact, the sector median forward P/S multiple is 3.6, so APLT would appear to be priced at a premium using those figures. However, I believe that the company’s revenues will likely compound relatively quickly upon receiving regulatory approvals.
Orphan drug costs are a concern. For example, orphan drugs have an average annual cost of $32,000, and more than a third of drugs with orphan indications cost more than $100,000 annually. (IQVIA, December 2020.)
For context, 1 in every 16,000 to 48,000 births are diagnosed with galactosemia. This means we can approximate the total population with this condition in the US at about 20,830 to 6,943. APLT’s corporate presentation suggests that in the US alone, there are about 3,000 patients with this condition, which, I believe, is a conservative estimate. Orphan-designated drugs typically cost an average of $32,000 per year, which would suggest a yearly US TAM of $666.6 million to $222.2 million. Hence, I believe APLT’s AT-007 (Govorestat) could quickly grow yearly sales beyond 2025.
Naturally, I don’t think it’s reasonable to assume it’ll reach 100% market share, but it does illustrate that there’s a lot of room for future growth post-approval. And that’s just from AT-007 for galactosemia. Plus, I used the 2020 average spending on orphan drugs as I couldn’t find a more recent figure, but it’s likely higher in 2024 due to inflation. Also, note that in 2020, a third of orphan drugs cost more than $100,000 per year, so my TAM estimate is likely very conservative.
Source: APLT Corporate Overview – March 2024.
Moreover, I estimate the company’s latest quarterly cash burn was roughly $18.9 million, which annualized implies a yearly cash burn of $75.6 million. I obtained these figures by adding APLT’s latest quarterly CFOs and Net CAPEX, then annualizing them. This suggests that APLT’s current cash runway is approximately 1.9 years, which seems more than enough time to see its first FDA approval. Once the company starts receiving product revenues, I expect its cash burn rate will decline as product sales offset it.
To give you an idea about this, APLT’s sector median gross margins are approximately 57.1%. So, I imagine that APLT will likely generate similar margins at the very least. If that’s the case, $100.0 million in yearly revenues would leave about $57.1 million in gross profit. Since APLT’s operating expenses for 2023 were just $20.6 million, I’d argue that around that sales figure would be enough to turn the company’s EBIT positive. Naturally, SG&A costs are likely to increase as APLT commercializes its products, but still, I believe there’s a wide margin to manouver and still generate positive EBIT. In this scenario, I doubt APLT would resort to further equity raises, as debt will likely be easier to secure and avoid shareholder dilution.
Consequently, I think it’s reasonable to say that the company has enough resources for the foreseeable future and that future dilution isn’t necessary at this stage, assuming there are no significant regulatory delays or setbacks. Therefore, I think APLT does seem like a compelling investment opportunity at these levels, with substantial upcoming catalysts in the near term and robust financials, which is why I rate it a “strong buy.”
Investment Caveats: Risk Analysis
Nevertheless, it’s worth noting that APLT is not without its risks. The main risk is regulatory, as there can still be FDA delays in the approval process, which would increase the delayed cashflows and decrease the company’s present value. Moreover, headline risk related to such regulatory setbacks would also likely impact the share price. And lastly, it’s important to consider that APLT’s execution risks related to commercialization efforts are also somewhat substantial. So far, the company seems competent in pushing its research through the FDA approval phases, but marketing and generating revenues are different challenges.
If management isn’t as skilled in navigating through insurance provider policies, healthcare provider relationships, and marketing the benefits of its drug candidate, then market acceptance could disappoint. Also, if the company fails to generate revenues with healthy margins, it’ll likely translate into a lower share price. Yet, I believe these risks are mostly justified by APLT’s clear pathway toward revenues and a wide TAM for its products, making it a viable investment for investors aware of the inherent biotech risks at this stage.
The recent pullback post-raise might offer a compelling entry price for new investors. (Source: TradingView.)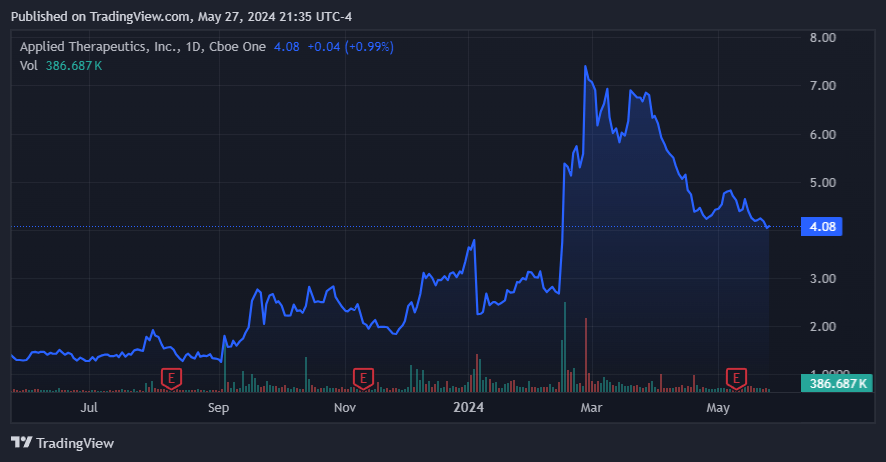
Strong Buy: Conclusion
Overall, APLT is a promising late-stage biotech company with several drug candidates nearing regulatory approval. Its leading drug candidate, AT-007 for galactosemia, will likely generate revenues by 2025, and other indications for the same drug will likely follow suit shortly after. Moreover, APLT’s balance sheet suggests the company has enough resources to fund its research until this happens. After that, I expect internally generated cash flows to extend its cash runway. Hence, despite the inherent biotech risks and potential commercialization pitfalls, the recent pullback might offer a compelling entry for new investors. Thus, I rate the stock a “strong buy” at these levels for those who understand the risks.

 Q2 2024 Earnings Call Transcript")

